
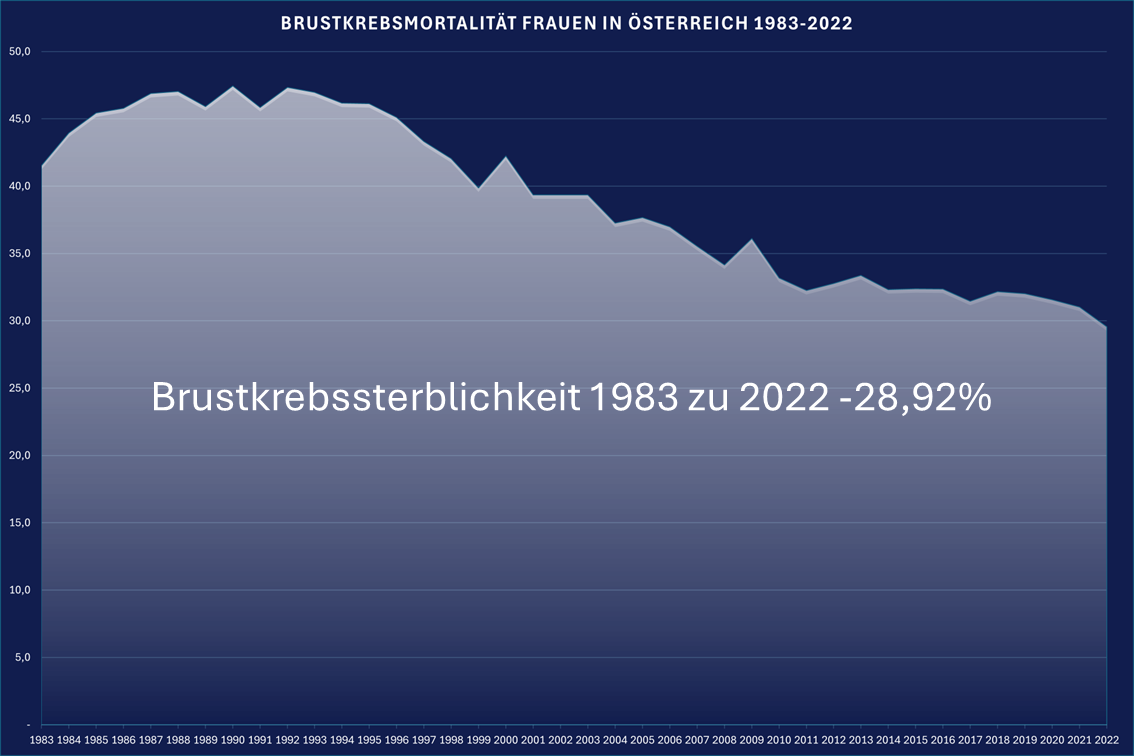
Dem medizinischen Fortschritt zu Folge konnte die Sterblichkeitsrate an Brustkrebs deutlich gesenkt werden. Starben im Jahr 1983 noch 41,5 von 100.000 Frauen waren es im Jahr 2022 um 28,92% weniger (Quelle Statistik Austria, Exceltabelle).
Neben der flächendeckenden Einführung von regelmäßigen Mammographien sind vor allem modernere Medikamente und Behandlungsstrategien sowie verbesserte chirurgischer Techniken für diese Entwicklung mitentscheidend.
Moderne Therapien können sich aber nur durch klinische Forschung entwickeln. Das Brustgesundheitszentrum der MedUni Wien ist hier Vorreiter, was klinische Studien auf dem Gebiet der Brustkrebsbehandlung betrifft. Dadurch ist es uns möglich, für unsere Patient:innen die modernsten Therapeutika einzusetzen.
- Studienpatient:innen helfen Therapien zu verbessern
- Studienpatient:innen leben länger
Durch die kontinuierliche Beobachtung über viele Jahre wird bei Studienpatient:innen eine Wiedererkrankung früher entdeckt und es werden auch frühzeitig andere Krankheiten erkannt.
Studien am Brustgesundheitszentrum
Gesunde Proband:innen
Eine randomisierte, doppelblinde, placebokontrollierte, multizentrische, internationale Phase-3-Studie zur Bestimmung der präventiven Wirkung von Denosumab auf Brustkrebs bei Frauen mit einer BRCA1-Keimbahnmutation.
- Frauen mit einer bestätigten schädlichen oder wahrscheinlich schädlichen BRCA-1-Keimbahnmutation (Variantenklasse 4 oder 5)
- Alter >= 25 Jahre und =< 55 Jahre bei Randomisierung
- Kein Nachweis von Brustkrebs durch MRT oder Mammographie (MG) und klinische Brustuntersuchung innerhalb der letzten 6 Monate vor der Randomisierung
- Kein klinischer Nachweis von Eierstockkrebs bei Randomisierung
- Negativer Schwangerschaftstest bei Randomisierung für Frauen im gebärfähigen Alter
- Zum Zeitpunkt der Randomisierung war keine vorbeugende Brustoperation geplant.
- Leistungsstatus 0 oder 1 der Eastern Cooperative Oncology Group (ECOG)
- Schriftliche Einverständniserklärung vor der Durchführung eines studienspezifischen Verfahrens
- Vorherige beidseitige Mastektomie
- Vorgeschichte von Eierstockkrebs (einschließlich Eileiter- und Bauchfellkrebs)
- Vorgeschichte von Brustkrebs
- Vorgeschichte eines invasiven Krebses mit Ausnahme von Basalzell- oder Plattenepithelkarzinom der Haut oder Carcinoma in situ des Gebärmutterhalses, papillärem oder follikulärem Schilddrüsenkrebs im Stadium 1, atypischer Hyperplasie oder LCIS (lobuläres Carcinoma in situ)
- Schwangere oder stillende Frauen (innerhalb der letzten 2 Monate vor der Randomisierung)
- Unwilligkeit, während und innerhalb von mindestens 5 Monaten nach Beendigung der Denosumab/Placebo-Therapie bei Frauen im gebärfähigen Alter eine hochwirksame Verhütungsmethode anzuwenden. (Hinweis: Frauen im gebärfähigen Alter sollten vor jeder Denosumab/Placebo-Injektion auf eine mögliche Schwangerschaft überwacht werden.)
-
Klinisch relevante Hypokalzämie (Anamnese und aktueller Zustand) oder Serumkalzium < 2,0 mmol/l (< 8,0 mg/dl)
-
Hypokalzämie definiert durch Kalzium unterhalb des Normalbereichs (ein einzelner Wert unterhalb des Normalbereichs stellt nicht unbedingt eine Hypokalzämie dar, sollte aber vor der Verabreichung der Dosis „korrigiert“ werden). Eine regelmäßige Überwachung des Kalziumspiegels (normalerweise vor der Verabreichung des Prüfpräparats [IP]) wird dringend empfohlen.
-
- Einnahme von Tamoxifen, Raloxifen oder Aromatasehemmern während der letzten 3 Monate vor der Randomisierung oder über einen Zeitraum von insgesamt mehr als 3 Jahren (aktuelle und vorherige Hormonersatztherapie [HRT] ist zulässig)
- Vorherige Anwendung von Denosumab
- Bei der Versuchsperson liegt eine bekannte Vorgeschichte oder ein aktueller Nachweis einer Osteonekrose oder Osteomyelitis des Kiefers vor oder es liegt eine aktive Zahn-/Kiefererkrankung vor, die innerhalb von 3 Monaten nach der Einschreibung eine orale Operation einschließlich Zahnextraktion erfordert.
- Gleichzeitige Behandlung mit einem Bisphosphonat oder einem anti-angiogenetischen Mittel
- Jegliche schwerwiegende medizinische oder psychiatrische Erkrankung, die den Teilnehmer daran hindern könnte, die Studie abzuschließen
- Bekannte aktive Infektion mit dem Hepatitis B-Virus oder dem Hepatitis C-Virus
- Bekannte Infektion mit dem humanen Immundefizienzvirus (HIV)
- Verwendung anderer Prüfpräparate (aktuelle oder frühere Einnahme von Aspirin oder nichtsteroidalen Antirheumatika [NSAR] ist zulässig)

Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at

DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Früherkennung mit Kontrastmittelmammographie: das VABABS-Projekt
Menschen mit dichtem Brustgewebe profitieren oft nur eingeschränkt von der konventionellen Mammografie, da die diagnostische Sensitivität (Anteil richtig erkannter Tumoren) in dieser Gruppe signifikant reduziert ist. Im Rahmen des VABABS-Projekts wird die Kontrastmittelmammografie (CEM) als potenziell effektive Methode zur Verbesserung der Brustkrebsfrüherkennung bei jener Risikogruppe evaluiert. CEM hat das Potenzial, die diagnostische Genauigkeit deutlich zu steigern und einen wesentlichen Beitrag zur Weiterentwicklung und Optimierung der Vorsorgestandards zu leisten.
Radiologische Brustambulanz – BGZ
Screening- und Assessmentzentrum des Österreichischen BrustkrebsfrüherkennungsprogrammesUniversitätsklinik für Radiologie und Nuklearmedizin
Leitstelle 7F
Montag bis Freitag: 7:30 bis 15:30 Uhr
Tel.: +43 (0)1 40400-48250 und 48240
Principal Investigator: Pascal Baltzer

Univ.-Prof. Priv.-Doz. Dr. Pascal A.T. Baltzer
Universitätsklinik für Radiologie und Nuklearmedizin
Währinger Gürtel 18-20
1090 Wien
E-Mail: pascal.baltzer@meduniwien.ac.at
Institution: Medical University of Vienna
Project title: Improvement and protection of women's health at a population level by Value Based Breast Screening in Vienna
Co-Principal Investigator(s): Eva Schernhammer (Medical University of Vienna) |
Natasa Peric (Medical University of Vienna)
Status: Ongoing (01.03.2023 – 28.02.2027)
Design: Prospektiv, multizentrisch, intra-individuell, populationsbasiert (Wien).
Förderung/Laufzeit: WWTF, 2023–2027.
Neoadjuvant (präoperativ)
ABCSG 61 / TEODOR Übersicht
Neoadjuvant TrEatment Optimization driven by ctDNA and endOcrine Responsiveness
Eine prospektive, randomisierte, offene, multizentrische Phase II Studie, die neoadjuvante endokrine Therapie, im Vergleich zu Chemotherapie, in Hormonrezeptor-positiven, HER2-negativen, ctDNA-negativen und endokrin sensitiven Patient:innen mit frühem und lokal fortgeschrittenem Brustkrebs untersucht
- Unterschriebene Einverständniserklärung, die vor allen studienspezifischen Beurteilungen und Verfahren eingeholt wurde
- Frauen und Männer ab 18 Jahren
-
Bei den Patientinnen muss ein histologisch bestätigter invasiver, einseitiger und lokal fortgeschrittener Brustkrebs mit den folgenden Merkmalen vorliegen:
- Stadium IIA-III gemäß AJCC (American Joint Committee on Cancer) Brustkrebs-Staging Version 8
- Histologisch bestätigte(r) hormonrezeptorpositive und HER2-negative(r) Tumor(en); die HER2-Messung erfolgt lokal gemäß den ASCO/CAP-Richtlinien. Bei multizentrischem und/oder multifokalem Tumor müssen alle histopathologisch untersuchten Tumoren die pathologischen Kriterien für hormonrezeptorpositive und HER2-negative Tumoren erfüllen.
- ER-positive Tumoren, d. h. >20 % positiv gefärbte Tumorzellen
- PR-positive oder -negative Tumoren
- Systemische Chemotherapie durch multidisziplinäres Tumorboard indiziert
- Fehlen einer vorherigen brustkrebsspezifischen Behandlung der aktuellen Malignität bei Beginn des Screenings
- Leistungsstatus der Eastern Cooperative Oncology Group (ECOG) von 0-2
-
Ausreichende Knochenmark- und Organfunktion, definiert durch die folgenden lokalen Laborwerte innerhalb von 8 Wochen vor Beginn der Studienbehandlung:
- Absolute Neutrophilenzahl (ANC) ≥ 1,5 × 109/l
- Thrombozyten ≥ 100 × 109/l
- Hämoglobin ≥ 10,0 g/dl
- Serumkreatinin innerhalb der normalen institutionellen Grenzen oder Kreatinin-Clearance ≥ 60 ml/min/1,73 m² für Patienten mit Serumkreatininwerten über dem institutionellen ULN.
- Alaninaminotransferase (ALT oder SGPT) ≤ 1,5 × Obergrenze des Normalwerts (ULN); Aspartataminotransferase (AST oder SGOT) ≤ 1,5 × ULN f. Gesamtserumbilirubin ≤ ULN; oder Gesamtbilirubin ≤ 3,0 × ULN mit direktem Bilirubin im Normalbereich bei Patienten mit gut dokumentiertem Gilbert-Syndrom
- Die Patienten müssen in der Lage und willens sein, orale Medikamente zu schlucken und bei sich zu behalten, ohne dass eine Erkrankung vorliegt, die die enterische Absorption beeinträchtigen würde.
- Der Patient muss bereit und in der Lage sein, die geplanten Besuche, Behandlungspläne, Labortests und anderen Studienverfahren einzuhalten.
- Bei Frauen im gebärfähigen Alter muss der Schwangerschaftstest im Urin oder Serum innerhalb der letzten 28 Tage vor der Registrierung negativ ausfallen. Bei postmenopausalen Frauen oder hysterektomierten Patientinnen müssen keine Schwangerschaftstests durchgeführt werden.
- Nicht geeignet für eine geeignete lokoregionale Behandlung (Brustoperation und/oder Strahlentherapie, falls angezeigt)
- Bilateraler invasiver Brustkrebs oder synchrones DCIS in der kontralateralen Brust
- Patienten, die während der Studie gleichzeitig eine systemische exogene Sexualhormontherapie erhalten (Hormonersatztherapie, orale oder andere hormonelle Kontrazeptiva wie eine hormonelle Verhütungsspirale usw.), sind nicht teilnahmeberechtigt, eine topische vaginale Östrogentherapie ist jedoch zulässig.
- Alle chronischen Medikamente, die für eine antineoplastische Behandlung kontraindiziert sind
- Teilnahme an einer vorherigen oder gleichzeitigen interventionellen Studie und Erhalt der Studienbehandlung (gleichzeitig oder innerhalb von 30 Tagen vor Behandlungsbeginn)
- Patienten, die zuvor eine systemische Krebstherapie gegen invasiven Brustkrebs erhalten haben
- Patienten mit einer Vorgeschichte von bösartigen Erkrankungen sind nicht teilnahmeberechtigt, außer unter den folgenden Umständen:
- Patienten mit einer anderen malignen Vorgeschichte als ausreichend behandeltem invasivem Brustkrebs sind geeignet, wenn sie seit mindestens 2 Jahren krankheitsfrei sind und vom Prüfarzt als sehr geringes Risiko für ein Wiederauftreten dieser malignen Erkrankung (z. B. Magenkrebs im Stadium I oder Hautkrebs) eingestuft werden.
- Patienten mit den folgenden Krebsarten sind teilnahmeberechtigt, auch wenn diese innerhalb der letzten 2 Jahre diagnostiziert und angemessen behandelt wurden: duktales Carcinoma in situ der Brust, Gebärmutterhalskrebs in situ und nicht metastasierter nichtmelanomatöser Hautkrebs
- Der Patient hat medizinische oder psychiatrische Störungen, die nach Einschätzung des Prüfarztes die Sicherheit des Patienten oder seine Einwilligung nach Aufklärung beeinträchtigen würden (z. B. bekannte unkontrollierte HIV-Infektion, chronische/aktive virale oder andere bekannte Hepatitis und/oder chronische Lebererkrankung, Zirrhose usw.).
- Unkontrollierte interkurrente Erkrankungen, einschließlich, aber nicht beschränkt auf, andauernde oder aktive Infektionen, symptomatische Herzinsuffizienz, instabile Angina Pectoris, Herzrhythmusstörungen, Diabetes oder psychiatrische Erkrankungen/soziale Situationen, die die Einhaltung der Studienanforderungen einschränken würden. Die Fähigkeit, die Studienanforderungen zu erfüllen, ist von jedem Prüfer zum Zeitpunkt der Auswahl für die Studienteilnahme zu beurteilen.
- Der Patient hat eine aktuelle Beeinträchtigung der gastrointestinalen (GI) Funktion oder eine GI-Erkrankung, die die Aufnahme der oralen Studienmedikamente signifikant verändern kann (z. B. unkontrollierte ulzerative Erkrankungen, unkontrollierte Übelkeit, Erbrechen oder Durchfall, Malabsorptionssyndrom oder Dünndarmresektion).
- Der Patient hat gleichzeitig eine andere schwere und/oder unkontrollierte Erkrankung, die nach Einschätzung des Prüfers inakzeptable Sicherheitsrisiken birgt, die Teilnahme des Patienten an der klinischen Studie kontraindiziert oder die Einhaltung des Protokolls gefährdet oder die Lebenserwartung auf ≤ 5 Jahre begrenzt.
- Schwangere oder stillende (stillende) Frauen oder Frauen, die während der Studienbehandlung und 6 Monate danach schwanger werden oder stillen möchten
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at

Dr.in Sabine Danzinger
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien

Assoc. Prof. Priv.-Doz. Dr. Georg Pfeiler
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
E-Mail: georg.pfeiler@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Operativ
Maßgeschneiderte Achselchirurgie mit oder ohne Achsellymphknotendissektion, gefolgt von Strahlentherapie bei Patienten mit klinisch lymphknotenpositivem Brustkrebs (TAXIS). Eine multizentrische randomisierte Phase-III-Studie (OPBC-03/ SAKK 23/16 /IBCSG 57-18 / ABCSG-53 / GBG-101)
Einschlusskriterien bei der Vorregistrierung:
- Schriftliche Einverständniserklärung gemäß ICH/GCP-Vorschriften vor allen studienspezifischen Verfahren.
- Brustkrebs, lymphknoten-positiv, nachgewiesen durch Palpation oder Bildgebung (mit oder ohne geplante neoadjuvante Behandlung)
- Weiblich oder männlich im Alter von ≥ 18 Jahren
- Fähigkeit, die Fragebögen zur Lebensqualität auszufüllen
Einschlusskriterien bei der Registrierung:
-
Lymphknotenpositiver Brustkrebs (histologisch oder zytologisch nachgewiesen sowohl im Primärtumor als auch im Lymphknoten) AJCC/UICC [42] Stadium II-III (alle molekularen Subtypen erlaubt):
- Durch Bildgebung (iN+) festgestellte und durch Pathologie bestätigte Knotenpositivität
- Durch Palpation festgestellte (cN1-3) und pathologisch bestätigte Knotenpositivität
- Okkulter Brustkrebs ist erlaubt, wenn durch Biopsie nachgewiesene Achsellymphadentastasen vorliegen
-
Geeignet für eine primäre ALND oder eine Sentinel-Lymphknoten-OP (SLN) mit Gefrierschnitt und entweder:
- Neu diagnostiziert
- Isoliertes Rezidiv in der Brust oder zweiter ipsilateraler Brustkrebs nach vorheriger brusterhaltender Operation und Sentinel-Prozedur und mindestens 3 Jahre krankheitsfrei und ohne vorherige Achseldissektion oder Achsel-RT
- Verdächtigster Achsellymphknoten geclippt
- Der Fragebogen zur Lebensqualität wurde ausgefüllt
- WHO-Leistungsstatus 0-2
- Ausreichende Voraussetzungen für Vollnarkose und Brustkrebsoperationen
- Frauen im gebärfähigen Alter verwenden eine wirksame Verhütungsmethode, sind weder schwanger noch stillen sie und verpflichten sich, während der Studienbehandlung und danach für den in den Leitlinien für adjuvante systemische Therapien empfohlenen Zeitraum nicht schwanger zu werden. Für alle Frauen im gebärfähigen Alter ist vor Aufnahme in die Studie ein negativer Schwangerschaftstest erforderlich.
- Männer verpflichten sich, während der Probebehandlung und sechs Monate danach kein Kind zu zeugen.
Einschlusskriterien bei Randomisierung (intraoperativ)
-
Lymphknotenpositiver Brustkrebs (histologisch oder zytologisch sowohl im Primärtumor als auch im Lymphknoten nachgewiesen) AJCC/UICC-Stadium II-III (alle molekularen Subtypen zulässig):
- Lymphknotenpositivität, die zunächst durch Bildgebung festgestellt wurde, und nicht tastbare und durch Pathologie** bestätigte Resterkrankung (einschließlich Rest-ITCs) bei SLN oder Nicht-SLN im Falle einer vorherigen neoadjuvanten Behandlung
-
Lymphknotenpositivität zunächst tastbar und Resterkrankung pathologisch bestätigt** (einschließlich Rest-ITCs) im Falle einer vorherigen neoadjuvanten Behandlung
- Hinweis: Patienten mit ypN0(i+) können eingeschlossen werden (das AJCC-Stadium II-III bezieht sich auf das Stadium vor der neoadjuvanten Behandlung). **Hinweis: Wenn die Feinnadelaspiration oder Stanzbiopsie des geclippten Lymphknotens nach der neoadjuvanten Behandlung eindeutig Krebs zeigt, ist eine wiederholte Bestätigung der Resterkrankung durch intraoperative Gefrierschnitte nicht zwingend erforderlich.
Ausschlusskriterien bei der Vorregistrierung:
Jeder potenzielle Patient, der eines der folgenden Kriterien erfüllt, muss von der Teilnahme an der Studie ausgeschlossen werden.
- Brustkrebs im Stadium IV
- Klinischer Brustkrebs N3c (klinischer N3a und klinischer N3b sind zulässig)
- Klinischer Brustkrebs N2b (klinischer N2a ist zulässig)
- Kontralateraler Brustkrebs innerhalb von 3 Jahren Hinweis: Kontralaterales duktales Carcinoma in situ (DCIS) ist zulässig, wenn die vorherige Behandlung die Studienbehandlung nicht beeinträchtigt oder beeinträchtigt.
- Vorherige Achseloperation (außer vorheriger Wächterlymphknoteneingriff im Falle eines Rezidivs in der Brust)
- Vorherige regionale Strahlentherapie
- Vorgeschichte einer malignen hämatologischen Erkrankung oder eines primären soliden Tumors, es sei denn, die Erkrankung ist seit der Vorregistrierung mindestens 3 Jahre lang in Remission, mit Ausnahme eines ausreichend behandelten zervikalen Carcinoma in situ oder eines lokalisierten nicht-melanozytären Hautkrebses.
- Behandlung mit einem experimentellen Medikament innerhalb von 30 Tagen nach der Vorregistrierung
- Jede andere schwerwiegende medizinische, psychiatrische, psychologische, familiäre oder geografische Erkrankung, die nach Einschätzung des Prüfarztes die geplante Inszenierung, Behandlung und Nachsorge beeinträchtigen, die Compliance des Patienten beeinflussen oder den Patienten einem hohen Risiko behandlungsbedingter Komplikationen aussetzen kann.
Ausschlusskriterien bei Randomisierung (intraoperativ):
Jeder potenzielle Patient, der eines der folgenden Kriterien erfüllt, muss von der Studie ausgeschlossen werden.
- Fehlen eines Clips im Probenröntgenbild
- Tastbare Erkrankung in der Achselhöhle nach maßgeschneiderter Achselchirurgie (TAS)
- Kein SLN in der Achselhöhle identifiziert

Assoc.Prof.in Priv.Doz.in Dr.in Ruth Exner
Stv. Leiterin des CCC-BrustgesundheitszentrumsUniversitätsklinik für Allgemeinchirurgie
Leiterin der Brustchirurgie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-56210
Tel. Patient:innenanmeldung: +43 (0)1 40400-64920
E-Mail: ruth.exner@meduniwien.ac.at
Studienbüro 9F
Sie erreichen das Studienbüro auf Ebene 9, Raum 9.F9.0, über die grünen Lifte.

Alesja Jona
(Karenz)Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43(0)1 40400-69950
E-Mail: alesja.jona@meduniwien.ac.at

Ildiko Velastin
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: Ildiko.velastin@meduniwien.ac.at

Michaela Urak
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: michaela.urak@meduniwien.ac.at
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at

Ass. Prof.in Dr.in Daphne Gschwantler-Kaulich
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Assoc. Prof. Priv.-Doz. Dr. Georg Pfeiler
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
E-Mail: georg.pfeiler@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Adjuvant (postoperativ)
EMBER-4: Eine randomisierte, offene Phase-3-Studie zum adjuvanten Imlunestrant im Vergleich zur standardmäßigen adjuvanten endokrinen Therapie bei Patientinnen, die zuvor 2 bis 5 Jahre lang eine adjuvante endokrine Therapie gegen ER+, HER2- Brustkrebs im Frühstadium mit erhöhtem Rezidivrisiko erhalten haben
- Sie haben die Diagnose ER+, HER2- Brustkrebs im Frühstadium, reseziert, invasiver Brustkrebs ohne Anzeichen von Fernmetastasen.
- Die Teilnehmer müssen ab dem Beginn der adjuvanten ET mindestens 24 Monate und nicht mehr als 60 Monate lang eine adjuvante ET erhalten haben.
- Die Teilnehmer haben möglicherweise eine (neo-)adjuvante Chemotherapie und/oder eine zielgerichtete Therapie mit einem CDK4/6- oder PARP-Inhibitor erhalten.
- Aufgrund klinisch-pathologischer Risikomerkmale muss ein erhöhtes Risiko für ein Wiederauftreten der Krankheit vorliegen.
- Haben Sie einen Leistungsstatus von 0 oder 1 auf der Skala der Eastern Cooperative Oncology Group.
- Verfügen über eine ausreichende Organfunktion.
- Liegen bei der primären Brustkrebsdiagnose irgendwelche Hinweise auf eine metastasierende Erkrankung (einschließlich kontralateraler ALN) oder einen entzündlichen Brustkrebs vor?
- Teilnehmer mit einer mehr als 6-monatigen Therapiepause im Verlauf der vorherigen adjuvanten ET.
- Teilnehmer, die die vorherige adjuvante ET mehr als 6 Monate vor dem Screening abgeschlossen oder abgebrochen haben.
- Teilnehmerinnen mit einer Vorgeschichte von Brustkrebs sind ausgeschlossen, mit Ausnahme von ipsilateralem DCIS, das vor ≥5 Jahren ausschließlich mit einer lokoregionären Therapie behandelt wurde.
- Schwangere, stillende Frauen oder Personen mit der Absicht, innerhalb der voraussichtlichen Dauer der Studie Kinder zu zeugen oder Kinder zu bekommen, beginnend mit dem Screeningbesuch bis 180 Tage nach der letzten Dosis der Studienintervention.
- Die Teilnehmerin hat zuvor ET beliebiger Dauer zur Brustkrebsprävention (Tamoxifen oder AIs) oder Raloxifen erhalten.
- Teilnehmer mit einer Vorgeschichte einer anderen Krebserkrankung.
- Sie leiden unter schwerwiegenden Vorerkrankungen, die nach Einschätzung des Prüfarztes eine Teilnahme an dieser Studie ausschließen würden.

Assoc.Prof. Priv.-Doz. Dr. Rupert Bartsch
Universitätsklinik für Innere Medizin I
Leiter der Brustambulanz der Onkologie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-44260
Tel. Patient:innenanmeldung: +43 (0)1 40400-44660
E-Mail: rupert.bartsch@meduniwien.ac.at
Heidrun Forstner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Beate Rottenmanner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Assoc. Prof. Priv.-Doz. Dr. Georg Pfeiler
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
E-Mail: georg.pfeiler@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Postneoadjuvante Phase-III-Studie zur Bewertung von Sacituzumab Govitecan, einem Antikörper-Wirkstoff-Konjugat bei Patientinnen mit primär HER2-negativem Brustkrebs und hohem Rückfallrisiko nach neoadjuvanter Standardbehandlung - SASCIA
- Vor Beginn bestimmter Protokollverfahren muss eine schriftliche Einverständniserklärung eingeholt und gemäß den örtlichen behördlichen Anforderungen dokumentiert werden. Dabei muss auch die erwartete Kooperation der Patienten bei der Behandlung und Nachsorge berücksichtigt werden.
- Alter bei Diagnose mindestens 18 Jahre.
- Bereitschaft und Fähigkeit, archivierte, in Formalin fixierte, in Paraffin eingebettete Gewebeblöcke (FFPE) aus der Operation nach neoadjuvanter Chemotherapie und aus der Kernbiopsie vor Beginn der neoadjuvanten Chemotherapie bereitzustellen, die zur zentralen prospektiven Bestätigung des HR-Status, des HER2-Status, des Ki-67 und der tumorinfiltrierenden Lymphozyten (TILs) sowie zur retrospektiven explorativen Korrelation zwischen Genen, Proteinen und mRNAs verwendet werden, die für die Empfindlichkeit/Resistenz gegenüber den Prüfpräparaten relevant sind. Bei Patienten mit bilateralem Karzinom müssen FFPE-Blöcke von beiden Seiten für die zentrale Untersuchung bereitgestellt werden.
- Histologisch bestätigtes einseitiges oder beidseitiges primäres invasives Mammakarzinom, histologisch bestätigt durch Kernbiopsie. Der Leittumor muss vom Prüfer anhand der Einschlusskriterien für den jeweiligen Subtyp und des Risikostatus definiert werden.
-
Zentral bestätigtes HER2-Negativ (IHC-Score 0-1 oder FISH-negativ gemäß ASCO/CAP-Richtlinie) und entweder
- HR-positive (≥1% positiv gefärbte Zellen) Krankheit oder
- HR-negativ (<1 % positiv gefärbte Zellen), vorzugsweise beurteilt anhand von Gewebe aus postneoadjuvanter residualer invasiver Erkrankung der Brust oder, falls nicht möglich, aus residualer Lymphknoteninvasion. Falls nicht auswertbar, wird der Kern einer diagnostischen Biopsie verwendet. Im Falle von bilateralem Brustkrebs muss der HER2-negative Status für beide Seiten bestätigt werden.
-
Patienten mit verbleibender invasiver Erkrankung nach neoadjuvanter Chemotherapie mit hohem Rezidivrisiko, definiert durch entweder:
- Für HR-negativ: jede verbleibende invasive Erkrankung > ypT1mi und/oder ypN1>1mm
- Bei HR-positiver Erkrankung: ein CPS+EG-Score ≥ 3 oder ein CPS+EG-Score von 2 und ypN+ unter Verwendung der lokalen ER und der Grad wurde anhand von Kernbiopsien ermittelt, die vor Beginn der neoadjuvanten Behandlung entnommen wurden.
- Angemessene chirurgische Behandlung einschließlich Resektion klinisch evidenter Erkrankungen und ipsilateraler Achsellymphknotendissektion. Von einer SNB vor NACT wird abgeraten. Eine Achseldissektion vor NACT ist nicht zulässig. Eine Achseldissektion, einschließlich gezielter Achseldissektion (TAD), sollte gemäß den Richtlinien durchgeführt werden. Eine histologische vollständige Resektion (R0) aller invasiven und in situ Tumoren ist erforderlich.
- Die Patienten müssen 16 Wochen lang eine neoadjuvante taxanhaltige Chemotherapie erhalten haben (Anthrazykline sind zulässig). In diesem Zeitraum müssen 6 Wochen einer taxanhaltigen neoadjuvanten Chemotherapie enthalten sein (Ausnahme: Bei Patienten mit progressiver Erkrankung, die nach mindestens 6-wöchiger taxanhaltiger neoadjuvanter Chemotherapie auftrat, ist auch eine Gesamtbehandlungsdauer von weniger als 16 Wochen zulässig).
- Keine klinischen Hinweise auf lokoregionale oder Fernrezidive während oder nach der präoperativen Chemotherapie. Lokale Progression während der Chemotherapie ist kein Ausschlusskriterium, wenn eine ausreichende lokale Kontrolle erreicht werden konnte.
- Im Falle einer lokalen Progression während der neoadjuvanten Therapie müssen vor Beginn der Studie Fernmetastasen durch geeignete Bildgebung (CT/MRT empfohlen) ausgeschlossen werden.
- Immuncheckpoint-Inhibitoren / Immuntherapien während der (neo)adjuvanten Therapie sind bis zum Abschluss der Strahlentherapie erlaubt.
- Patienten mit bekannter gBRCA1/2-Mutation ohne Indikation zur adjuvanten Olaparib-Therapie dürfen an der Studie teilnehmen.
- Es muss ein Zeitraum von weniger als 16 Wochen seit dem Datum der letzten Operation oder von weniger als 10 Wochen seit dem Abschluss der Strahlentherapie (je nachdem, was zuletzt eintritt) und dem Datum der Randomisierung eingehalten werden.
- Die Strahlentherapie sollte vor Beginn der Studienbehandlung durchgeführt werden. Eine Strahlentherapie der Brust ist bei allen Patientinnen mit brusterhaltender Operation und der Brustwand und Lymphknoten gemäß den lokalen Richtlinien sowie bei allen Patientinnen mit cT3/4- oder ypN+-Krankheit, die durch Mastektomie behandelt wurden, angezeigt.
- Leistungsstatus 0 oder 1 der Eastern Cooperative Oncology Group (ECOG).
- Abklingen aller akuten toxischen Wirkungen einer vorherigen Krebstherapie oder eines chirurgischen Eingriffs oder einer Strahlentherapie auf NCI CTCAE v 5.0 Grad ≤ 1 (ausgenommen Alopezie oder andere Toxizitäten, die nach Ermessen des Prüfers nicht als Sicherheitsrisiko für den Patienten betrachtet werden).
- Geschätzte Lebenserwartung von mindestens 5 Jahren, unabhängig von der Diagnose Brustkrebs.
- Der Patient muss für geplante Besuche, Behandlungen und Nachuntersuchungen erreichbar sein.
- Eine normale Herzfunktion nach neoadjuvanter Chemotherapie muss gemäß den lokalen Richtlinien bestätigt werden. Die Ergebnisse für LVEF müssen über dem normalen Grenzwert der Einrichtung liegen.
-
Laboranforderungen:
Hämatologie
- Absolute Neutrophilenzahl (ANC) ≥1,5 x 109 / L
- Thrombozyten ≥100 x 109 / l
- Hämoglobin ≥10 g/dL (≥6,2 mmol/L) Leberfunktion
- Gesamtbilirubin <1,25 x UNL
- AST und ALT ≤ 1,5 x UNL
- Alkalische Phosphatase ≤2,5x UNL Nierenfunktion
- <1,25 x ULN-Kreatinin oder Kreatinin-Clearance ≥ 30 ml/min (laut Cockroft-Gault, wenn Kreatinin über UNL liegt).
-
Negativer Schwangerschaftstest (Urin oder Serum) innerhalb von 14 Tagen vor der Randomisierung für alle Frauen im gebärfähigen Alter. Eine Frau gilt als gebärfähig, wenn sie nicht postmenopausal ist. Postmenopausal ist definiert als:
- Alter ≥60 Jahre
- Alter <60 Jahre und ≥12 Monate ununterbrochene Amenorrhoe ohne erkennbare Ursache außer der Menopause
- Chirurgische Sterilisation (bilaterale Oophorektomie und/oder Hysterektomie).
- Für Frauen im gebärfähigen Alter und Männer mit Partnern im gebärfähigen Alter: Zustimmung zur Abstinenz (Verzicht auf heterosexuellen Geschlechtsverkehr) oder zur Anwendung von Verhütungsmethoden mit einer Versagerquote von < 1 % pro Jahr während des Behandlungszeitraums und für mindestens 6 Monate nach der letzten Dosis Sacituzumab Govitecan bei weiblichen Patienten und für mindestens 3 Monate bei männlichen Patienten; für mindestens 6 Monate nach der letzten Dosis Capecitabin oder Carboplatin/Cisplatin bei weiblichen Patienten und für mindestens 3 Monate nach der letzten Dosis Capecitabin oder 6 Monate nach der letzten Dosis Carboplatin/Cisplatin bei männlichen Patienten. Beispiele für nicht-hormonelle Verhütungsmethoden mit einer Versagerquote von < 1 % pro Jahr sind: beidseitige Tubenligatur; Sterilisation des männlichen Partners; Intrauterinpessare.
- Vollständige Staging-Untersuchung vor Einleitung der neoadjuvanten Chemotherapie. Fehlende Staging-Untersuchungen müssen vor der Randomisierung durchgeführt werden.
- Bekannte Überempfindlichkeitsreaktion auf eine der in diesem Protokoll verwendeten Verbindungen oder Substanzen.
- Patienten mit eindeutigen klinischen oder radiologischen Hinweisen auf Krebs im Stadium IV (metastasierte Erkrankung) sind nicht teilnahmeberechtigt.
- Patienten mit bekannter gBRCA1/2-Mutation und angezeigter oder geplanter adjuvanter Olaparib-Therapie, sofern verfügbar.
-
Patienten mit einer malignen Erkrankung in der Vorgeschichte sind mit folgenden Ausnahmen nicht teilnahmeberechtigt:
- Der Patient ist seit mindestens 5 Jahren krankheitsfrei und hat ein geringes Risiko für ein Wiederauftreten der bösartigen Erkrankung
- CIS des Gebärmutterhalses, Basalzell- und Plattenepithelkarzinome der Haut.
- Patientinnen: Schwangerschaft oder Stillzeit zum Zeitpunkt der Randomisierung oder Absicht, während der Studie schwanger zu werden und bis zu 6 Monate nach Sacituzumab Govitecan und bis zu 6 Monate nach der Behandlung mit Capecitabin oder Carboplatin/Cisplatin.
-
Schwerwiegende und relevante Komorbiditäten, die mit der Anwendung zytotoxischer Mittel oder der Teilnahme an der Studie interagieren würden, einschließlich Morbus Gilbert, Crigler-Najjar-Syndrom, bekannte Hepatitis B, Hepatitis C, bekannte HIV-Positivität oder bekannte Autoimmunerkrankungen außer Diabetes, Vitiligo oder stabile Schilddrüsenerkrankung, Vitiligo oder andere Autoimmunerkrankungen der Haut mit dermatologischen Manifestationen sind nur zulässig, sofern alle der folgenden Bedingungen erfüllt sind:
- Der Ausschlag muss < 10 % der Körperoberfläche bedecken
- Die Krankheit ist zu Beginn gut unter Kontrolle und erfordert nur niedrig dosierte topische Kortikosteroide
- Kein Auftreten akuter Exazerbationen der Grunderkrankung, die Psoralen plus Ultraviolett-A-Strahlung (PUVA), Methotrexat, Retinoide, biologische Wirkstoffe, orale Calcineurin-Inhibitoren oder hochwirksame oder orale Kortikosteroide innerhalb der letzten 12 Monate erfordert hätten.
- Jeder Zustand, der die sichere Verabreichung der vom Arzt gewählten Behandlung beeinträchtigt, falls der Patient zufällig dem TPC-Arm zugeteilt wird.
- Bekannte oder vermutete kongestive Herzinsuffizienz (> NYHA I) und/oder koronare Herzkrankheit, Angina Pectoris, die eine antianginöse Medikation erfordert, früherer Herzinfarkt in der Anamnese, Hinweise auf einen früheren Infarkt im EKG, nicht oder schlecht kontrollierte arterielle Hypertonie (d. h. Blutdruck > 150/90 mmHg unter Behandlung mit maximal drei blutdrucksenkenden Medikamenten), Rhythmusstörungen, die eine dauerhafte Behandlung erfordern (ausgenommen chronisches Vorhofflimmern, für das kein Herzschrittmacher erforderlich ist), klinisch signifikante Klappenerkrankung des Herzens, supraventrikuläre und nodale Arrhythmien, die einen Herzschrittmacher erfordern oder nicht mit Medikamenten kontrolliert werden können; Reizleitungsstörung, die einen Herzschrittmacher erfordert.
- Vorgeschichte einer idiopathischen Lungenfibrose, einer organisierenden Pneumonie (z. B. Bronchiolitis obliterans), einer medikamenteninduzierten Pneumonitis, einer idiopathischen Pneumonitis oder einer aktiven Pneumonitis im CT-Thorax-Scan.
- Erhalt eines Lebendimpfstoffs innerhalb von 30 Tagen vor Studienbeginn oder innerhalb von 30 Tagen nach Erhalt der Chemotherapie.
- Vorgeschichte schwerwiegender neurologischer oder psychiatrischer Störungen, einschließlich psychotischer Störungen, Demenz oder Krampfanfälle, die das Verständnis und die Erteilung einer informierten Einwilligung verhindern würden.
- Jeder Zustand, der nach Ansicht des Prüfers die Bewertung der Studienbehandlung oder die Interpretation der Patientensicherheit oder der Studienergebnisse beeinträchtigen würde.
- Bekannte allergische Reaktionen auf Irinotecan.
-
Gleichzeitige Behandlung mit:
- Chronische Kortikosteroide vor Studienbeginn, mit Ausnahme von intranasalen und inhalierten Kortikosteroiden oder systemischen Kortikosteroiden in physiologischen Dosen, die 10 mg/Tag Prednison oder ein gleichwertiges Kortikosteroid nicht überschreiten dürfen.
Assoc.Prof. Priv.-Doz. Dr. Rupert Bartsch
Universitätsklinik für Innere Medizin I
Leiter der Brustambulanz der Onkologie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-44260
Tel. Patient:innenanmeldung: +43 (0)1 40400-44660
E-Mail: rupert.bartsch@meduniwien.ac.at
Heidrun Forstner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Beate Rottenmanner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Eine offene, randomisierte Phase-III-Studie zur Bewertung der Wirksamkeit und Sicherheit einer erweiterten Therapie mit Camizestrant (AZD9833, ein selektiver oraler Östrogenrezeptor-Degrader der nächsten Generation) im Vergleich zur Standard-Endokrintherapie (Aromatasehemmer oder Tamoxifen) bei Patienten mit ER+/HER2- frühem Brustkrebs und mittlerem oder hohem Rezidivrisiko, die eine definitive lokoregionale Therapie und mindestens 2 Jahre standardmäßige adjuvante endokrine Therapie ohne Rezidiv abgeschlossen haben
- Frauen und Männer, ≥18 Jahre zum Zeitpunkt des Screenings (oder gemäß nationalen Richtlinien)
- Histologisch bestätigter ER+/HER2- resezierter invasiver Brustkrebs im Frühstadium mit hohem oder mittlerem Rezidivrisiko, basierend auf klinisch-pathologischen Risikomerkmalen, wie im Protokoll definiert.
- Abgeschlossene adäquate (definitive) lokoregionale Therapie (Operation mit oder ohne Strahlentherapie) des/der primären Brusttumors/-tumoren, mit oder ohne (neo)adjuvante Chemotherapie
- Mindestens 2 Jahre, aber nicht mehr als 5 Jahre (+3 Monate) adjuvante ET (+/- CDK4/6-Inhibitor) abgeschlossen
- Leistungsstatus der Eastern Cooperative Oncology Group (ECOG) von ≤ 1
- Ausreichende Organ- und Knochenmarkfunktion
- Inoperabler lokal fortgeschrittener oder metastasierter Brustkrebs
- Pathologische Komplettremission nach Behandlung mit neoadjuvanter Therapie
- Eine andere Krebserkrankung in der Anamnese (außer nicht-melanozytärem Hautkrebs oder Carcinoma in situ des Gebärmutterhalses oder eine Erkrankung mit sehr geringem Rezidivrisiko nach Einschätzung des Prüfarztes), es sei denn, sie ist seit mindestens 5 Jahren ab dem Zeitpunkt der Randomisierung in vollständiger Remission ohne Therapie.
- Jegliche Hinweise auf schwere oder unkontrollierte systemische Erkrankungen, die nach Ansicht des Prüfers eine Teilnahme an der Studie oder eine Compliance ausschließen
- Bekannte LVEF <50 % mit Herzinsuffizienz NYHA-Grad ≥2.
- Mittleres Ruhe-QTcF-Intervall >480 ms beim Screening
- Gleichzeitige exogene Reproduktionshormontherapie oder nicht-topische Hormontherapie bei nicht krebsbedingten Erkrankungen
- Jede gleichzeitige Krebsbehandlung, die nicht im Protokoll angegeben ist, mit Ausnahme von Bisphosphonaten (z. B. Zoledronsäure) oder RANKL-Inhibitoren (z. B. Denosumab)
- Vorherige Behandlung mit Camizestrant, experimentellen SERDs/erprobten ER-Zielsubstanzen oder Fulvestrant
- Derzeit schwanger (bestätigt durch positiven Serum-Schwangerschaftstest) oder stillend
- Patienten mit bekannter Überempfindlichkeit gegen aktive oder inaktive Hilfsstoffe von Camizestrant oder Arzneimitteln mit ähnlicher chemischer Struktur oder Klasse wie Camizestrant. Bei prä-/perimenopausalen weiblichen und männlichen Patienten bekannte Überempfindlichkeit oder Unverträglichkeit gegenüber LHRH-Agonisten, die den Patienten von der Verabreichung eines LHRH-Agonisten ausschließen würde
Assoc.Prof.in Priv.Doz.in Dr.in Ruth Exner
Stv. Leiterin des CCC-BrustgesundheitszentrumsUniversitätsklinik für Allgemeinchirurgie
Leiterin der Brustchirurgie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-56210
Tel. Patient:innenanmeldung: +43 (0)1 40400-64920
E-Mail: ruth.exner@meduniwien.ac.at
Studienbüro 9F
Sie erreichen das Studienbüro auf Ebene 9, Raum 9.F9.0, über die grünen Lifte.
Alesja Jona
(Karenz)Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43(0)1 40400-69950
E-Mail: alesja.jona@meduniwien.ac.at
Ildiko Velastin
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: Ildiko.velastin@meduniwien.ac.at
Michaela Urak
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: michaela.urak@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Eine offene, randomisierte Phase-III-Studie zur Bewertung der Wirksamkeit und Sicherheit von Camizestrant (AZD9833, ein selektiver oraler Östrogenrezeptor-Degrader der nächsten Generation) im Vergleich zur Standard-Endokrintherapie (Aromatasehemmer oder Tamoxifen) als adjuvante Behandlung für Patienten mit ER+/HER2- frühem Brustkrebs und einem mittelhohen oder hohen Rezidivrisiko, die die endgültige lokoregionale Behandlung abgeschlossen haben und keine Anzeichen einer Erkrankung aufweisen
- Frauen und Männer; ≥18 Jahre zum Zeitpunkt des Screenings (oder gemäß nationalen Richtlinien)
- Histologisch bestätigter ER+/HER2- resezierter invasiver Brustkrebs im Frühstadium ohne jegliche Hinweise auf eine Metastasierung, wie im Protokoll definiert.
- Abgeschlossene adäquate (endgültige) lokoregionale Therapie (Operation mit oder ohne Strahlentherapie) für den/die primären Brusttumor(e), mit oder ohne (neo)adjuvante Chemotherapie.
- Die Patientinnen müssen innerhalb von 12 Monaten nach der endgültigen Brustoperation randomisiert werden.
- Die Patientinnen können vor der Randomisierung bis zu 12 Wochen lang eine endokrine Therapie erhalten haben.
- Leistungsstatus der Eastern Cooperative Oncology Group (ECOG) von ≤ 1
- Ausreichende Organ- und Knochenmarkfunktion
- Inoperabler lokal fortgeschrittener oder metastasierter Brustkrebs
- Pathologische Komplettremission nach Behandlung mit neoadjuvanter Therapie
- Eine andere Krebserkrankung in der Anamnese (außer nicht-melanozytärem Hautkrebs oder Carcinoma in situ des Gebärmutterhalses oder eine Erkrankung mit sehr geringem Rezidivrisiko nach Einschätzung des Prüfarztes), es sei denn, sie ist seit mindestens 5 Jahren ab dem Zeitpunkt der Randomisierung in vollständiger Remission ohne Therapie.
- Jegliche Hinweise auf schwere oder unkontrollierte systemische Erkrankungen, die nach Ansicht des Prüfers eine Teilnahme an der Studie oder eine Compliance ausschließen"
- Bekannte LVEF <50 % mit Herzinsuffizienz NYHA-Grad ≥2.
- Mittleres Ruhe-QTcF-Intervall > 480 ms beim Screening
- Gleichzeitige exogene Reproduktionshormontherapie oder nicht-topische Hormontherapie bei nicht krebsbedingten Erkrankungen
- Jede gleichzeitige Krebsbehandlung, die nicht im Protokoll angegeben ist, mit Ausnahme von Bisphosphonaten (z. B. Zoledronsäure) oder RANKL-Inhibitoren (z. B. Denosumab)
- Vorherige Behandlung mit Camizestrant, experimentellen SERDs/erprobten ER-Zielsubstanzen oder Fulvestrant
- Derzeit schwanger (bestätigt durch positiven Serum-Schwangerschaftstest) oder stillend.
- Patienten mit bekannter Überempfindlichkeit gegen aktive oder inaktive Bestandteile von Camizestrant oder Arzneimittel mit ähnlicher chemischer Struktur oder Klasse wie Camizestrant. Bei prä-/perimenopausalen weiblichen und männlichen Patienten bekannte Überempfindlichkeit oder Unverträglichkeit gegenüber LHRH-Agonisten, die den Patienten von der Verabreichung eines LHRH-Agonisten ausschließen würde.
Assoc.Prof.in Priv.Doz.in Dr.in Ruth Exner
Stv. Leiterin des CCC-BrustgesundheitszentrumsUniversitätsklinik für Allgemeinchirurgie
Leiterin der Brustchirurgie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-56210
Tel. Patient:innenanmeldung: +43 (0)1 40400-64920
E-Mail: ruth.exner@meduniwien.ac.at
Studienbüro 9F
Sie erreichen das Studienbüro auf Ebene 9, Raum 9.F9.0, über die grünen Lifte.
Alesja Jona
(Karenz)Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43(0)1 40400-69950
E-Mail: alesja.jona@meduniwien.ac.at
Ildiko Velastin
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: Ildiko.velastin@meduniwien.ac.at
Michaela Urak
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: michaela.urak@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Metastasiert
Offene, randomisierte Phase-3-Studie zum Vergleich von Gedatolisib in Kombination mit Fulvestrant und mit oder ohne Palbociclib mit Standardtherapien bei Patienten mit HR-positivem, HER2-negativem fortgeschrittenem Brustkrebs, die zuvor mit einem CDK4/6-Inhibitor in Kombination mit einer nichtsteroidalen Aromatasehemmer-Therapie behandelt wurden
- Histologisch oder zytologisch bestätigte Diagnose von metastasiertem oder lokal fortgeschrittenem Brustkrebs. Erwachsene Frauen, prä- und/oder postmenopausal, und erwachsene Männer. Prämenopausale (und perimenopausale) Frauen können aufgenommen werden, wenn sie für eine Behandlung mit einem LHRH-Agonisten geeignet sind. Die Patienten müssen vor oder an Zyklus 1, Tag 1 mit einer gleichzeitigen Behandlung mit einem LHRH-Agonisten begonnen haben und bereit sein, diese für die Dauer der Studie fortzusetzen.
- Negativer Schwangerschaftstest für Frauen im gebärfähigen Alter. Weibliche Probanden im gebärfähigen Alter müssen vom Screening bis 1 Jahr nach der letzten Dosis der Studienbehandlung eine wirksame und/oder akzeptable Verhütungsmethode anwenden.
- Bestätigte Diagnose eines positiven Östrogenrezeptors und/oder positiven Progesteronrezeptors gemäß den Leitlinien der American Society of Clinical Oncology/College of American Pathologists (ASCO-CAP) (2020), basierend auf der letzten Tumorbiopsie unter Verwendung eines Tests, der den lokalen Standards entspricht
- Dokumentierte HER2-Immunhistochemie (IHC) negativ gemäß ASCO-CAP-Leitfaden 2018
- Ausreichendes Archiv- oder frisches Tumorgewebe für die Analyse des PIK3CA-Mutationsstatus
- Der Patient muss eine Dokumentation des radiologischen Krankheitsverlaufs während oder nach der letzten Vorbehandlung haben und außerdem eine radiologisch bewertbare Krankheit (messbar und/oder nicht messbar) gemäß RECIST v1.1 haben, gemäß lokaler Beurteilung. Patienten mit einer ausschließlich knochenbezogenen Erkrankung müssen lytische oder gemischte lytische/blastische Läsionen haben, die genau beurteilt werden können; ausschließlich knochenbezogene blastische Läsionen ohne Weichteilanteil sind nicht zulässig.
- Leistungsstatus der Eastern Cooperative Oncology Group (ECOG) von 0-1
- Lebenserwartung von mindestens 3 Monaten
- Fortschreiten während oder nach einer Kombinationsbehandlung mit CDK4/6-Inhibitoren und nichtsteroidalen Aromatasehemmern (AI)
- Ausreichende Knochenmarks-, Leber-, Nieren- und Gerinnungsfunktion
- Anamnese mit anderen malignen Erkrankungen als ausreichend behandeltem nicht-melanozytärem Hautkrebs, kurativ behandeltem in situ-Gebärmutterhalskrebs oder anderen soliden Tumoren, die kurativ behandelt wurden und bei denen seit mindestens 3 Jahren keine Krankheitsanzeichen vorliegen
- Vorherige Behandlung mit einem Phosphoinositid-3-Kinase (PI3K)-Inhibitor, einem Proteinkinase-B-Inhibitor (Akt) oder einem mechanistischen Ziel von Rapamycin (mTOR)-Inhibitor
- Eine vorherige Behandlung mit Chemotherapie und Antikörper-Wirkstoff-Konjugaten bei fortgeschrittener Erkrankung ist nicht zulässig (eine vorherige adjuvante oder neoadjuvante Chemotherapie ist zulässig)
- Mehr als 2 Linien vorheriger endokriner Therapiebehandlung
- Reine Knochenerkrankung, die ausschließlich blastisch ist und keine Weichteilkomponente aufweist
- Personen mit Typ-1-Diabetes oder unkontrolliertem Typ-2-Diabetes
-
Bekannte und unbehandelte oder aktive Hirn- oder leptomeningeale Metastasen
a. Probanden mit vorbehandelten Metastasen im zentralen Nervensystem (ZNS) können in die Studie aufgenommen werden, wenn sie die folgenden Kriterien erfüllen: Sie benötigen keine unterstützende Therapie mit Steroiden, haben keine Anfälle und zeigen keine unkontrollierten neurologischen Symptome, die Krankheit ist stabil und wurde durch eine Röntgenuntersuchung innerhalb von mindestens 4 Wochen vor der Aufnahme bestätigt.
- Patienten mit fortgeschrittener, symptomatischer, viszeraler Ausbreitung, bei denen das Risiko lebensbedrohlicher Komplikationen kurzfristig besteht
-
Klinisch signifikante kardiovaskuläre Anomalien in der Anamnese, wie z. B.: Kongestive Herzinsuffizienz (Klassifikation der New York Heart Association (NYHA) ≥ II) innerhalb von 6 Monaten vor Studienbeginn
- Herzinfarkt innerhalb von 12 Monaten nach Studienbeginn
- Vorgeschichte jeglicher unkontrollierter (oder unbehandelter) klinisch signifikanter Herzrhythmusstörungen (z. B. ventrikuläre Tachykardie), kompletter Linksschenkelblock, hochgradiger AV-Block (z. B. bifaszikulärer Block, Mobitz Typ II und AV-Block dritten Grades), supraventrikulärer oder nodaler Arrhythmien oder Reizleitungsstörungen in den letzten 12 Monaten
- Unkontrollierte Hypertonie, definiert durch systolischen Blutdruck (SBP) ≥160 mmHg und/oder diastolischen Blutdruck (DBP) ≥100 mmHg, mit oder ohne blutdrucksenkende Medikamente (die Einleitung oder Anpassung der Einnahme blutdrucksenkender Medikamente ist vor dem Screening zulässig)
-
Langes QT-Syndrom, idiopathischer plötzlicher Herztod oder angeborenes langes QT-Syndrom in der Familienanamnese oder eines der folgenden:
- i. Risikofaktoren für Torsades de Pointes (TdP), einschließlich nicht korrigierter Hypokaliämie oder Hypomagnesiämie oder klinisch signifikanter/symptomatischer Bradykardie in der Vorgeschichte
- ii. Beim Screening konnte das korrigierte QT-Intervall mithilfe der Fridericia-Formel (QTcF) im EKG nicht bestimmt werden (d. h. nicht lesbar oder nicht interpretierbar) oder QTcF > 480 ms (beim Screening anhand von dreifachen EKGs ermittelt)
- Bekannte Überempfindlichkeit gegen die Studienmedikamente oder deren Bestandteile
- Schwangere oder stillende Frauen
-
Gleichzeitige Teilnahme an einer anderen interventionellen klinischen Studie
- Die Probanden müssen zustimmen, während ihrer Teilnahme an VIKTORIA-1 zu keinem Zeitpunkt an einer anderen klinischen Studie (mit Ausnahme von Beobachtungsstudien) teilzunehmen.
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Eine randomisierte, doppelblinde Phase-3-Studie mit Tucatinib oder Placebo in Kombination mit Trastuzumab und Pertuzumab als Erhaltungstherapie bei metastasiertem HER2+-Brustkrebs (HER2CLIMB-05)
- Zentral bestätigtes HER2+-Brustkarzinom gemäß den Leitlinien des College of American Pathologists (CAP) der American Society of Clinical Oncologists (ASCO) von 2018 vor der Randomisierung (definiert als ein Wert von 3+ in der Immunhistochemie (IHC) und/oder 2+ IHC und gleichzeitig positiv durch ISH).
-
Sie leiden an einer nicht resektablen, lokal fortgeschrittenen oder metastasierten Erkrankung.
- Bei einem Wiederauftreten (nach [neo]adjuvanter Therapie) muss eine mindestens 6-monatige Behandlungspause von Trastuzumab und Pertuzumab im Rahmen einer fortgeschrittenen HER2+-Erkrankung im Frühstadium von Brustkrebs eingehalten werden.
- Vor Studienbeginn haben die Teilnehmer 4-8 Zyklen einer Induktionstherapie erhalten, die ausschließlich Trastuzumab, Pertuzumab und Taxan als Erstlinientherapie zur Behandlung von fortgeschrittenem Brustkrebs enthielt. Teilnehmer sind berechtigt, sofern sie nach Abschluss der Induktionstherapie keine Anzeichen eines Fortschreitens der Krankheit aufweisen.
- Bekannter Hormonrezeptorstatus (gemäß lokalen Richtlinien; kann hormonrezeptorpositiv [HR+] oder -negativ [HR-] sein)
- Leistungsstatus der Eastern Cooperative Oncology Group (ECOG) von 0 oder 1
-
Einbeziehung des ZNS - Basierend auf einer kontrastmittelverstärkten Magnetresonanztomographie (MRT) des Gehirns können die Teilnehmer Folgendes aufweisen:
- Keine Hinweise auf Hirnmetastasen
- Unbehandelte Hirnmetastasen, die asymptomatisch sind und keiner sofortigen lokalen Behandlung bedürfen und, sofern sie bei früheren Hirnbildgebungsverfahren identifiziert wurden, seit Beginn der Erstlinien-Induktionstherapie mit Trastuzumab, Pertuzumab und Taxan keine Anzeichen einer Progression aufweisen.
-
Zuvor behandelte Hirnmetastasen, die asymptomatisch sind
- Hirnmetastasen, die zuvor mit lokaler Therapie behandelt wurden, dürfen seit der Behandlung nicht weiter fortgeschritten sein.
- Vorherige Behandlung mit einem Tyrosinkinasehemmer, der auf HER2 und/oder den epidermalen Wachstumsfaktor-Rezeptor (EGFR) abzielt, einschließlich Pyrotinib, Lapatinib, Tucatinib, Neratinib und Afatinib (außer Neratinib, wenn es in einem erweiterten adjuvanten Setting verabreicht wird und seit der letzten Neratinib-Dosis vor Beginn der Studienmedikation ≥ 12 Monate vergangen sind)
- Keine Möglichkeit, sich einer kontrastmittelverstärkten MRT des Gehirns zu unterziehen
-
Ausschluss des ZNS – basierend auf einer MRT-Untersuchung des Gehirns und einer klinischen Beurteilung
- Symptomatische Hirnmetastasen nach ZNS-gerichteter lokaler Therapie
- Fortschreiten von Hirnmetastasen seit Beginn der Erstlinientherapie mit Trastuzumab, Pertuzumab und Taxan
- Fortlaufende Anwendung systemischer Kortikosteroide mit einer täglichen Gesamtdosis von >2 mg Dexamethason (oder Äquivalent)
- Jede unbehandelte Hirnverletzung an einer anatomischen Stelle, die ein Risiko für den Teilnehmer darstellen könnte
- Bekannte oder vermutete leptomeningeale Erkrankung (LMD)
- Schlecht kontrollierte (> 1/Woche) Anfälle oder andere anhaltende neurologische Symptome
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Registerstudien
Dieses Register ist eine prospektive und retrospektive, multizentrische Sammlung von Daten über Patientinnen mit metastasiertem Brustkrebs in Österreich. Alle Tumormerkmale, Krankengeschichten und auch Behandlungssequenzen werden in anonymisierter Form dokumentiert. Für die Dokumentation im Register sind keine weiteren diagnostischen oder therapeutischen Maßnahmen erforderlich als jene, die ohnehin generell notwendig sind. Die Teilnahme am Register darf die Behandlungsabläufe nicht beeinträchtigen. Vor der Eingabe von Daten muss eine schriftliche Einwilligung eingeholt werden. Bei verstorbenen Patienten ist keine Einverständniserklärung erforderlich.
Assoc.Prof. Priv.-Doz. Dr. Rupert Bartsch
Universitätsklinik für Innere Medizin I
Leiter der Brustambulanz der Onkologie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-44260
Tel. Patient:innenanmeldung: +43 (0)1 40400-44660
E-Mail: rupert.bartsch@meduniwien.ac.at
Heidrun Forstner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Beate Rottenmanner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at

Biobanken stellen eine elementare Voraussetzung für die onkologische Forschung dar.
Um die Sammlung von Blutproben voranzutreiben, wurde 2021 die CCC-BGZ Biobank ins Leben gerufen.
Wir danken unseren Partnern für ihre Unterstützung.

Dr.in Christine Deutschmann
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
H2O - National Health Outcomes Observatory
Auftraggeberin: MedUni Wien
Laufzeit: fortlaufend seit 2022
Ansprechperson GÖG: Alexander Degelsegger-Márquez
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Die ReCare-Studie ist eine europäische Registerstudie, in die Patient:innen eingeschlossen werden können, die erneut eine Bestrahlung wegen Lokalrezidiven, neuem Primär- oder Sekundärkrebs erhalten.
- Pathologisch bestätigter Krebs.
- Schriftliche Einverständniserklärung für E²-RADIatE gemäß den geltenden rechtlichen und ethischen Anforderungen
- Geplanter Einsatz der Strahlentherapie
- Die Eignung des Patienten für mindestens eine bestätigte Kohorte
- Patienten ab 12 Jahren

OÄ.in Dr.in Daniela Kauer Dorner
Leiterin Tumorgruppe MammakarzinomUniversitätsklinik für Radioonkologie
Währinger Gürtel 18-20
1090 Wien

Dr. Stefan Konrad
Universitätsklinik für Radioonkologie
Währinger Gürtel 18-20
1090 Wien
E-Mail: stefan.konrad@meduniwien.ac.at
Validierung des Xpert® Insight Breast Cancer Tests zur Stratifizierung der Ergebnisse bei prämenopausalen Frauen mit Hormonrezeptor-positivem, HER2-negativem Brustkrebs im Frühstadium und Bewertung der Übereinstimmung von Xpert® Breast Cancer STRAT4 mit der IHC-Bewertung von Routine-Biomarkern für Brustkrebs (ER, PgR, HER2 und Ki67) - eine retrospektive translationale Studie der Biomarker-Kohorte ABCSG-12
Assoc.Prof.in Priv.Doz.in Dr.in Ruth Exner
Stv. Leiterin des CCC-BrustgesundheitszentrumsUniversitätsklinik für Allgemeinchirurgie
Leiterin der Brustchirurgie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-56210
Tel. Patient:innenanmeldung: +43 (0)1 40400-64920
E-Mail: ruth.exner@meduniwien.ac.at
Studienbüro 9F
Sie erreichen das Studienbüro auf Ebene 9, Raum 9.F9.0, über die grünen Lifte.
Alesja Jona
(Karenz)Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43(0)1 40400-69950
E-Mail: alesja.jona@meduniwien.ac.at
Ildiko Velastin
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: Ildiko.velastin@meduniwien.ac.at
Michaela Urak
Universitätsklinik für Allgemeinchirurgie
Studienbüro Brustchirurgie
Tel.: +43 (0)1 40400-24620
E-Mail: michaela.urak@meduniwien.ac.at
Assoc. Prof. Priv.-Doz. Dr. Georg Pfeiler
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
E-Mail: georg.pfeiler@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Nicht interventionelle Studien
Eine nicht-interventionelle, einarmige, multizentrische Studie mit ambispektivem Studiendesign zur Beschreibung von PatientInnen mit HER2 positivem Brustkrebs, die neoadjuvant behandelt werden, und Entwicklung einer dynamischen Risikobewertung, um das Risiko von Fernmetastasen vorherzusagen.
- Histologisch bestätigtes Adenokarzinom der Brust
- HER2+ Erkrankung definiert als 3+ durch Immunhistochemie oder 2+ durch Immunhistochemie und mit HER2-Amplifikation durch In-Situ-Hybridisierung (ISH)
- Patients mit bestätigtem ein- / beidseitigem Brustkrebs kann eingeschlossen werden, wenn mindestens eine Läsion HER2+ ist
- Prospective Patienten: die mindestens eine Dosis eines neoadjuvanten Behandlungsschemas mit dualer HER2-Blockade erhalten haben oder für die mindestens eine Dosis einer neoadjuvanten Behandlung mit dualer HER2-Blockade vorgesehen ist, falls die Patientinnen vor Beginn der neoadjuvanten Therapie zugestimmt haben
- Retrospective Patientinnen: die mindestens eine Dosis eines neoadjuvanten Behandlungsschemas mit dualer HER2-Blockade erhalten haben, gefolgt von einer definitiven Operation.
- Beginn der Anti-HER2-Therapie max. 5 Jahre vor der Registrierung
- Metastasierte oder lokal fortgeschrittene Erkrankung (ohne lokoregionale Behandlungsmöglichkeiten mit kurativer Absicht) zum Zeitpunkt der Diagnose
- Frühere Krebstherapie für dieselbe Erkrankung, außer der in dieser Nicht-interventionellen Studie (NIS) bewerteten Therapie
Assoc.Prof. Priv.-Doz. Dr. Rupert Bartsch
Universitätsklinik für Innere Medizin I
Leiter der Brustambulanz der Onkologie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-44260
Tel. Patient:innenanmeldung: +43 (0)1 40400-44660
E-Mail: rupert.bartsch@meduniwien.ac.at
Heidrun Forstner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Beate Rottenmanner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Univ.-Prof. Dr. Christian Singer
Leiter des CCC-BrustgesundheitszentrumsUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-28010
Tel. Patient:innenanmeldung: +43 (0)1 40400-28040
E-Mail: christian.singer@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
PEDRO („Patient Experience Data in Radiation Oncology“): ein Pilot Projekt zur Erhebung von „Real-world-data“ aus Patient:innenperspektive.
- alle Patienten, die eine externe Strahlentherapie erhalten
- nicht in der Lage, Fragebögen zu strahleninduzierten Nebenwirkungen selbstständig zu beantworten
OÄ.in Dr.in Daniela Kauer Dorner
Leiterin Tumorgruppe MammakarzinomUniversitätsklinik für Radioonkologie
Währinger Gürtel 18-20
1090 Wien
Dr. Stefan Konrad
Universitätsklinik für Radioonkologie
Währinger Gürtel 18-20
1090 Wien
E-Mail: stefan.konrad@meduniwien.ac.at
Eine epidemiologische, prospektive Kohortenstudie zur Generierung von Real-World-Evidenz bei Patienten mit fortgeschrittenem HR+/HER2-Brust Krebs, behandelt in der Erstliniensituation nach aktuellem Behandlungsstandard mit einer endokrinen Kombinationstherapie mit Palbociclib
- Nachweis einer persönlich unterzeichneten und datierten Einverständniserklärung, aus der hervorgeht, dass der Patient über alle relevanten Aspekte der Studie informiert wurde.
- Diagnose von HR+/HER2- lokal fortgeschrittenem, inoperablem oder metastasiertem Brustkrebs.
- Der Arzt hat entschieden, dass eine Erstlinienbehandlung mit Palbociclib (i) in Kombination mit einem Aromatasehemmer oder (ii) in Kombination mit Fulvestrant bei Frauen angezeigt ist, die zuvor eine endokrine Therapie gemäß der aktuellen lokalen Produktkennzeichnung erhalten haben. Bei Frauen vor oder in der Perimenopause sollte die endokrine Therapie mit einem Agonisten des luteinisierenden Hormon-Releasing-Hormons (LHRH) kombiniert werden.
- Patienten, die nach Ansicht des Prüfers bereit und in der Lage sind, regelmäßige Klinikbesuche gemäß dem örtlichen Behandlungsstandard am Studienort durchzuführen.
- Alter 18 Jahre oder älter.
- Alle Kontraindikationen gemäß der aktuellen lokalen Produktkennzeichnung.
- Vorherige systemische antineoplastische Behandlung bei fortgeschrittener Erkrankung. Ausnahme: Der Beginn der Erstlinienbehandlung mit Palbociclib in Kombination mit Aromatasehemmer oder Fulvestrant gemäß der aktuellen lokalen Produktkennzeichnung ist bis zu 4 Wochen vor Aufnahme zulässig.
- Patienten, die zum Zeitpunkt der Aufnahme an einer interventionellen klinischen Studie teilnehmen, die Prüfpräparate oder bereits vermarktete Produkte umfasst. Hinweis: Eine gleichzeitige Teilnahme an anderen nicht-interventionellen/beobachtenden Studien, Registern und translationalen Forschungsnetzwerken (z. B. PRAEGNANT, OPAL) oder Akteneinsichten ist zulässig.
- Patienten, die den Zweck der Studie nicht verstehen oder nicht bereit sind, eine Einverständniserklärung zu unterzeichnen.
Assoc.Prof. Priv.-Doz. Dr. Rupert Bartsch
Universitätsklinik für Innere Medizin I
Leiter der Brustambulanz der Onkologie
Währinger Gürtel 18-20
A-1090 Wien
Tel.: +43 (0)1 40400-44260
Tel. Patient:innenanmeldung: +43 (0)1 40400-44660
E-Mail: rupert.bartsch@meduniwien.ac.at
Heidrun Forstner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Beate Rottenmanner
Universitätsklinik für Innere Medizin I
Währinger Gürtel 18-20
A-1090 Wien
Assoc. Prof. Priv.-Doz. Dr. Georg Pfeiler
Universitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
E-Mail: georg.pfeiler@meduniwien.ac.at
DGKP Ingeborg Brandl, MSc
Head Study Nurse, Breast Care NurseUniversitätsklinik für Frauenheilkunde
Währinger Gürtel 18-20
A-1090 Wien
Tel. Sekretariat Studienzentrum: 01-40400 79870
E-Mail: ingeborg.brandl@meduniwien.ac.at
Translationale Forschung / Präzisionsmedizin

Im Christian Doppler Labor für patient:innenzentrierte Brustbildgebung der MedUni Wien beschäftigen sich Forscher:innen mit der Entwicklung und Anwendung von Diagnosetools, die nicht nur präziser sind als derzeitige Methoden, sondern auch angenehmer für die Patient:innen. Damit soll die Akzeptanz der mitunter lebensrettenden Vorsorgeuntersuchungen weiter erhöht werden.
Technische Fortschritte in der Brustbildgebung haben sich in den letzten Jahrzehnten hauptsächlich auf die Lösung des medizinischen Problems der diagnostischen Genauigkeit konzentriert. Gängige bildgebende Verfahren werden von vielen Personen als unangenehm, ja sogar schmerzhaft, empfunden, mit ein Grund für die nach wie vor mangelhafte Akzeptanz des Brustkrebs-Screenings.

Dieses CD-Labor unter der Leitung Prof. Baltzer von der Universitätklinik für Radiologie und Nukleearmedizin verfolgt daher einen Triple-S (Soft, Safe und Smart) Ansatz: Sicherstellen des Patient:innenwohls (Soft), eine Erhöhung der Akzeptanz und Wirksamkeit (Safe) der Bildgebung und die Einbindung von grundlegenden Innovationen (Smart).
Radiologische Brustambulanz – BGZ
Screening- und Assessmentzentrum des Österreichischen BrustkrebsfrüherkennungsprogrammesUniversitätsklinik für Radiologie und Nuklearmedizin
Leitstelle 7F
Montag bis Freitag: 7:30 bis 15:30 Uhr
Tel.: +43 (0)1 40400-48250 und 48240

Team: Georg Langs, Yen Tan, Christoph Bock, Paulina Gebhart, Ivana Janícková, Thomas Helbich, Bettina Roethlin, Valentin Militevic, Zsuzsanna Bago-Horvath, Katja Pinker, Christian Singer, Martin Ortner
PREDICTOME untersucht, ob sich mit modernen Bildgebungen (PET/MRT) und Blut-/Gewebedaten früh vorhersagen lässt, wie gut eine neoadjuvante Therapie wirkt. Die Behandlung selbst bleibt unverändert – wir analysieren nur zusätzliche Informationen, um zukünftige Therapien besser auf einzelne Patient:innen abzustimmen.
PREDICTOME ist ein interdisziplinäres Forschungsprojekt, das vom Wissenschafts- und Technologiefonds Wien (WWTF) finanziert wird.
Publikationen
- Fürböck, C., Perkonigg, M., Helbich, T., Pinker, K., Romeo, V. and Langs, G., 2022, September. Identifying Phenotypic Concepts Discriminating Molecular Breast Cancer Sub-Types. In Medical Image Computing and Computer Assisted Intervention–MICCAI 2022: 25th International Conference, Singapore, September 18–22, 2022, Proceedings, Part VII(pp. 276-286).
- Romeo, V., Helbich, T.H. and Pinker, K., 2022. Breast PET/MRI Hybrid Imaging and Targeted Tracers. Journal of Magnetic Resonance Imaging.